Wernicke Korsakoff Sendromu (WKS), tiamin (B1 vitamini) eksikliği nedeniyle ortaya çıkan bir bozukluktur. Wernicke ensefalopatisi olarak da bilinen Wernicke Korsakoff sendromu, üç ana klinik semptomla karakterize edilen nörolojik bir hastalıktır.
Haber Merkezi / Konfüzyon, istemli hareketi koordine edememe (ataksi) ve göz (oküler) anormallikleri. Wernicke sendromu, WKS’nin akut fazı olarak kabul edilir ve tedavi edilmezse kronik geri dönüşümsüz Korsakoff sendromuna geçiş yapar. Bu iki bozukluk birlikte ortaya çıktığında Wernicke-Korsakoff sendromu terimi kullanılır.
Belirtileri ve semptomları
WKS, tedavi edilmediği takdirde kronik geri dönüşümsüz Korsakoff sendromuna (Wernicke sendromu) yol açan tiamin eksikliğinin bir sonucudur. Her ikisinden de semptomlar örtüşen hastalara WKS teşhisi konur.
Wernicke sendromu, üç ana klinik semptomla karakterize edilir: zihinsel durum değişiklikleri, istemli hareketi koordine edememe (ataksi) ve göz anormallikleri. Etkilenen bireyler üç semptomu da göstermeyebilir.
Wernicke sendromuna bağlı konfüzyon ve oryantasyon bozukluğu birkaç gün veya hafta içinde gelişir ve bozuklukla ilişkili ana problemdir. Tiamin eksikliği ile ortaya çıkan akut bir sendromdur. Etkilenen bireyler uyuşukluk, dikkatsizlik, uyuşukluk ve ilgisizlik yaşayabilir.
Deliryum, özellikle alkolü de bırakan alkolik hastalarda sıklıkla görülür. Tedavi edilmezse, etkilenen kişilerde stupor veya bilinç kaybı (koma) gelişebilir. Etkilenen bazı kişilerde yavaş, dengesiz bir yürüyüş olabilir. Hastalığın akut aşamasında bu, etkilenen kişinin yardım almadan ayakta durmasını veya yürümesini engelleyebilir.
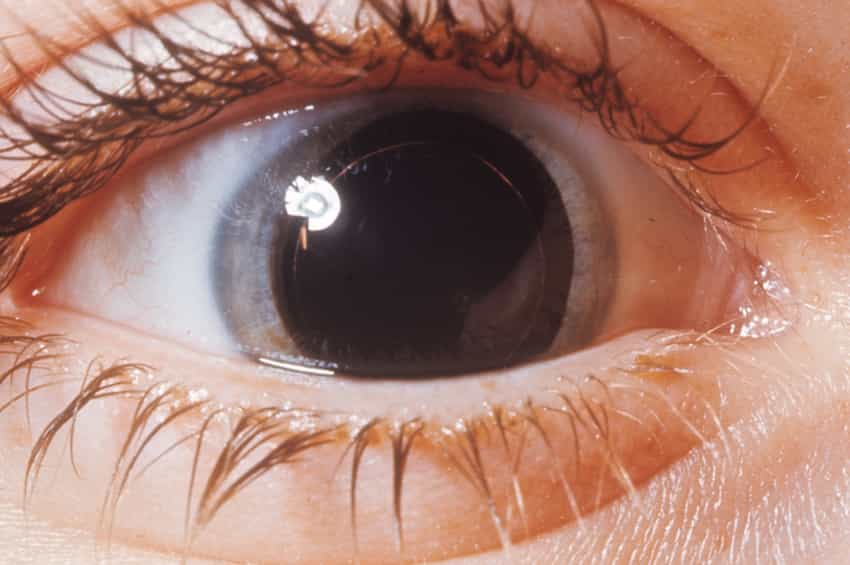
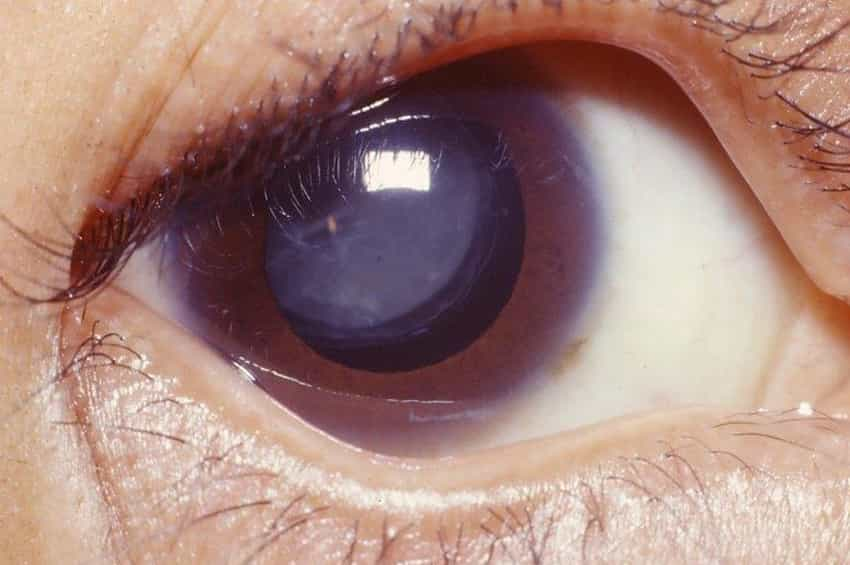
Wernicke sendromu ile ilişkili oküler anormallikler arasında çift görme, hızlı, istemsiz göz hareketleri (nistagmus), bazı göz kaslarının felci (oftalmopleji) ve nadiren üst göz kapaklarının sarkması (pitoz) yer alır.
Wernicke sendromlu bireylerin yaklaşık yüzde 80-90’ında Korsakoff sendromu gelişir. Korsakoff sendromunun semptomları genellikle Wernicke sendromunun zihinsel semptomları azalmaya başladığında gelişir.
Korsakoff sendromu, hafıza bozukluğu, özellikle kısa süreli hafıza kaybı (yani, yeni hatıralar oluşturamama veya yeni bilgileri muhafaza edememe) ile karakterize edilir. Etkilenen bazı bireylerde uzun süreli hafızaların rastgele kaybı da olabilir. Nadiren, bireyler hafızalarındaki boşlukları doldurmak için hayali olaylar yaratabilirler (konfabulasyon).
Dikkat ve sosyal davranış nispeten korunur. Etkilenen insanlar, normal görünebilecek sosyal olarak uygun bir konuşmayı sürdürebilirler. Korsakoff sendromlu kişiler genellikle hastalıklarının farkında değildir.
WKS uzun süreli veya kalıcı olabilir ve birçok siniri (polinöropati), özellikle merkezi sinir sistemi dışındakileri (periferik nöropati) etkiler. Periferik nöropati, kol ve bacaklarda güçsüzlüğe neden olabilir ve yürüme güçlüğüne katkıda bulunabilir.
WKS’li bireylerde hızlı kalp atışı (taşikardi), ayakta dururken düşük kan basıncı (postural hipotansiyon) ve bilinç kaybı (senkop) dahil olmak üzere çeşitli kardiyovasküler anormallikler de ortaya çıkabilir. Hipotermi (vücudun çok hızlı ısı kaybetmesi) da WKS’nin bir belirtisi olabilir.
Diğer semptomlar uzuvlarda zayıflık, zayıf kas koordinasyonu, dengesiz yürüyüş, yavaş yürüme, hızlı göz hareketleri, göz kaslarında felç, zayıf ince motor fonksiyon ve azalmış koku alma duyusunu içerebilir.
WKS’nin akut fazının ileri evreleri, hastaların %10-20’sinde komaya ve ölüme yol açar.
Nedenleri
WKS, tiamin (B1 vitamini) eksikliğinden kaynaklanır. Tiamin, beyin tarafından enerji için kullanılan glikozun metabolize edilmesi için gerekli olan önemli bir besindir.
Tiamin eksikliği, sıcaklığı, iştahı, duyguları ve büyümeyi düzenleyen hipotalamusun aktivitesi dahil, metabolik olarak en aktif beyin bölgelerindeki beyin fonksiyonlarını etkiler. Tiamin eksikliği, sinir ve kardiyovasküler sistem hücrelerini diğer organ sistemlerinin hücrelerine göre daha fazla etkiler.
Tanısı
WKS tanısı kapsamlı bir klinik değerlendirme ve ayrıntılı bir hasta öyküsü temelinde konur. Rutin laboratuvar taramaları ve karaciğer fonksiyon testleri gibi testler, benzer sunumlara sahip diğer bozuklukları ekarte edebilir.
Tiamin ve eritrosit transketolaz aktivitesini (her ikisi de WKS’de azalır) ölçen testler de tanıya ulaşmada yardımcı olabilir. Tümörleri, enfarktüsleri ve kanamayı (hemoraji) dışlamak için bilgisayarlı tomografi (BT) taraması ve manyetik rezonans görüntüleme (MRI) gerekli olabilir.
BT taramaları ve MRG’ler ayrıca WKS’nin göstergesi olan beyin değişikliklerini de ortaya çıkarabilir. Memeli cisimler, beyinde bulunan ve limbik sistemin bir parçasını oluşturan bir çift küçük yuvarlak yapıdır. Limbik sistem, beyinde duygularla ve hafıza oluşumuyla ilgili bir grup yapıdır.
Tedavisi
WKS’li bireyleri tedavi etmek için derhal tiamin uygulaması (replasman tedavisi) kullanılır. Tiamin intravenöz olarak verilir, çünkü etkilenen bireylerde abdominal absorpsiyon engellenebilir. Mental durum değişiklikleri, görme anormallikleri ve ataksi genellikle tiamin uygulamasıyla düzelir. Tiamin uygulaması birkaç ay boyunca günlük olarak devam edebilir. Erken tedavi ile Wernicke sendromu mutlaka WKS’ye ilerlemeyecektir.
Alkolden uzak durma ve uygun diyet değişiklikleri önerilir. WKS’li kişilerde magnezyum ve potasyum genellikle düşüktür. Bu ve diğer elektrolitlerin tiamine ek olarak eklenmesi gerekebilir. Kronik olarak yetersiz beslenen hastalar, normal alıma devam edilene kadar tüm B vitaminlerinin takviyesini almalıdır. Hastanın iyileşme düzeyine bağlı olarak multidisipliner bir ekip gerekebilir.
Hasta semptomlarını yönetmek için nöroloji, psikiyatri (akıl sağlığı), oftalmoloji (göz doktoru), kardiyoloji (kalp) ve gastroenteroloji (mide ve bağırsak sorunları) uzmanlarına ihtiyaç duyulabilir. Ayrıca, etkilenen bazı kişiler, zihinsel ve duygusal bozuklukları tedavi etmek için tasarlanmış psikolojik yöntemlerden (psikoterapi) yararlanabilir.
Hastaların hareket ve yürüyüşte yaşadıkları zorluklar fizik tedavi ile tedavi edilebilir. Yürüme güçlükleri, başlangıçtaki hareket kaybına ve tedavinin zamanlamasına bağlı olarak kalıcı olabilir.
Bozukluk erken yakalanırsa ve tedaviye hemen başlanırsa, tam veya önemli bir iyileşme sağlanabilir. Bazı hastalarda kafa karışıklığı ve zihinsel sorunların çözülmesi aylar alabilir. Şiddetli vakalarda beyin hasarı, hafıza ve yürüme ile ilgili kalıcı sorunlara neden olabilir.
Erken müdahaleye rağmen, WKS’li hastalar tamamen iyileşemeyebilir ve uzun süreli rehabilitasyon ve desteğe ihtiyaç duyabilir. Diğer tedavi semptomatik ve destekleyicidir.
Not: Sunulan bilgilerin amacı herhangi bir hastalığı teşhis veya tedavi etmek, iyileştirmek veya önlemek değildir. Tüm bilgiler yalnızca genel bilginize yöneliktir, tıbbi tavsiye veya belirli tıbbi durumların tedavisinin yerine geçmez. Uygulamadan önce bu bilgileri doktorunuzla görüşün.