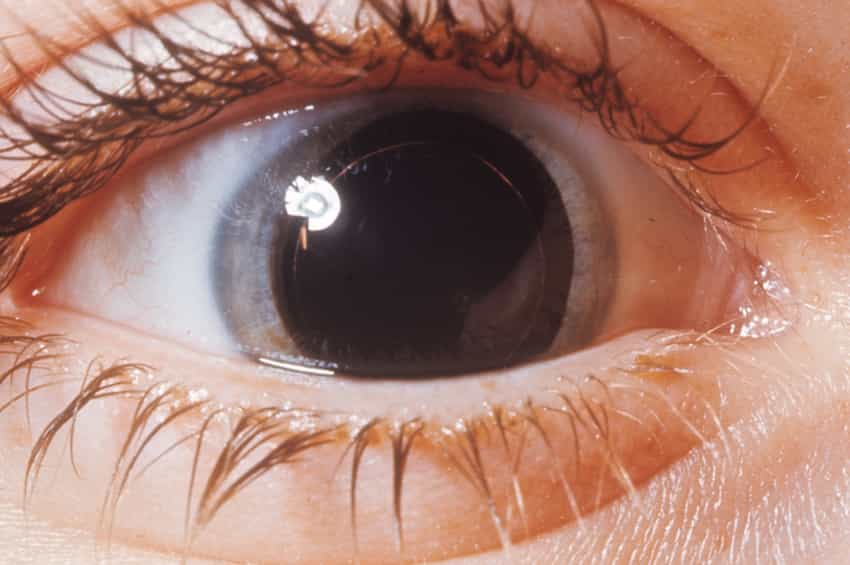
Weill Marchesani sendromu, göz merceği anormallikleri, boy kısalığı, alışılmadık derecede kısa, geniş kafa (brakisefali) ve eklem sertliği ile karakterize nadir bir genetik bağ dokusu bozukluğudur.
Haber Merkezi / Göz (oküler) anormallikleri arasında küçük yuvarlak mercekler (mikrosferofaki), merceğin anormal konumu (ektopi lentis), gözün ve merceğin anormal şeklinden kaynaklanan miyopi (miyopi) ve optik sinire zarar veren göz hastalığı (glokom) yer alabilir. Etkilenen bazı bireylerde kalp kusurları mevcuttur. Weill Marchesani sendromu, otozomal resesif veya otozomal dominant kalıtımı izler.
Belirtileri ve semptomları
Weill-Marchesani sendromu ile ilişkili semptom ve bulgular kişiden kişiye değişir. Weill-Marchesani sendromu, göz merceği anormallikleri, kısa boy, alışılmadık derecede kısa, geniş kafa (brakisefali) ve eklem sertliği ile karakterizedir. Etkilenen birçok kişide, dar bir ağız çatısı (damak) dahil olmak üzere ek kraniyofasiyal anormallikler vardır; küçük, az gelişmiş bir üst çene (maksiller hipoplazi); ve/veya belirli dişlerin şekil bozukluğu ve yanlış hizalanması.
Etkilenen kişilerde genellikle, normalde lensleri yerinde tutmaya yardımcı olan belirli liflerin (zonula ciliaris) kısmen veya tamamen yokluğuyla birlikte mikrosferofakya (normalden daha küçük ve yuvarlak bir lens) vardır. Sonuç olarak, bozukluğu olan bazı kişilerde lenslerin progresif olarak yerinden çıkması (ektopia lentis) gelişmeye yatkın olabilir veya bu duruma doğumda sahip olabilirler (konjenital lentis ektopisi).
Ektopia lentis, lenslerin kayması veya eğilmesi (yani, kısmi yer değiştirme veya subluksasyon) veya tam dislokasyon (lüksasyon) ile karakterize edilebilir; gözler (iridodonez). Ek oküler anormallikler de Weill-Marchesani sendromu ile ilişkilendirilebilir. Bunlar arasında göz merceğinin şeffaflığının kaybı (katarakt); Aköz hümör olarak bilinen ince, sulu sıvıyı içeren gözün renkli bölgelerinin (iris) önündeki bölmelerin (yani ön kamaraların) anormal sığlığı; ve/veya sekonder glokom. Glokom, göz sıvısının anormal derecede artan basıncı ile karakterizedir.
Weill-Marchesani sendromlu bireyler, azalan görüş netliği ve netliği (keskinlik), belirgin miyopluk (miyopi) veya körlük dahil olmak üzere değişen derecelerde görme bozukluğuna sahip olabilir. Görme bozukluğunun derecesi, mevcut göz anormalliklerinin ciddiyetine ve/veya kombinasyonuna bağlıdır. Glokom, göz sıvısının anormal derecede artan basıncı ile karakterizedir. Weill-Marchesani sendromlu bireyler, azalan görüş netliği ve netliği (keskinlik), belirgin miyopluk (miyopi) veya körlük dahil olmak üzere değişen derecelerde görme bozukluğuna sahip olabilir.
Görme bozukluğunun derecesi, mevcut göz anormalliklerinin ciddiyetine ve/veya kombinasyonuna bağlıdır. Glokom, göz sıvısının anormal derecede artan basıncı ile karakterizedir. Weill-Marchesani sendromlu bireyler, azalan görüş netliği ve netliği (keskinlik), belirgin miyopluk (miyopi) veya körlük dahil olmak üzere değişen derecelerde görme bozukluğuna sahip olabilir. Görme bozukluğunun derecesi, mevcut göz anormalliklerinin ciddiyetine ve/veya kombinasyonuna bağlıdır.
Boy kısalığı genellikle mevcuttur ve parmaklar kısa olabilir. Ek olarak, bazı kişilerde, özellikle ellerde olmak üzere belirli eklemlerde ilerleyici sertlik gelişebilir. Nadiren kalp anormallikleri bildirilmiştir ve bunlar arasında aort ile pulmoner arter arasında bir açıklığın kaldığı bir kusur (patent duktus arteriyozis), daralmış bir pulmoner kapak (pulmoner stenoz) ve yakın zamanda torasik aort anevrizması ve servikal arter diseksiyonu yer alır.
Nedenlerı
Weill Marchesani sendromu, otozomal resesif veya otozomal dominant kalıtımı izler.
Resesif genetik bozukluklar, bir birey her bir ebeveynden anormal bir gen miras aldığında ortaya çıkar. Bir birey, hastalık için bir normal gen ve bir anormal gen alırsa, kişi hastalığın taşıyıcısı olur, ancak genellikle semptom göstermez. Taşıyıcı iki ebeveynin her ikisinin de anormal geni geçirme ve dolayısıyla etkilenen bir çocuğa sahip olma riski her hamilelikte %25’tir. Ebeveynler gibi taşıyıcı olan bir çocuğa sahip olma riski her hamilelikte %50’dir. Bir çocuğun her iki ebeveynden de normal gen alma şansı %25’tir. Risk erkekler ve kadınlar için aynıdır.
ADAMTS10 geninin otozomal resesif Weill Marchesani sendromu ile ilişkili olduğu bulunmuştur.
Baskın genetik bozukluklar, belirli bir hastalığa neden olmak için anormal bir genin yalnızca tek bir kopyası gerektiğinde ortaya çıkar. Anormal gen, her iki ebeveynden de kalıtsal olabilir veya etkilenen bireyde mutasyona uğramış (değişmiş) bir genin sonucu olabilir. Anormal genin etkilenen bir ebeveynden bir yavruya geçme riski her gebelik için %50’dir. Risk erkekler ve kadınlar için aynıdır.
FBN1 ve LTBP2 genlerinin, her bir ailede otozomal dominant Weill Marchesani sendromu ile ilişkili olduğu bulunmuştur.
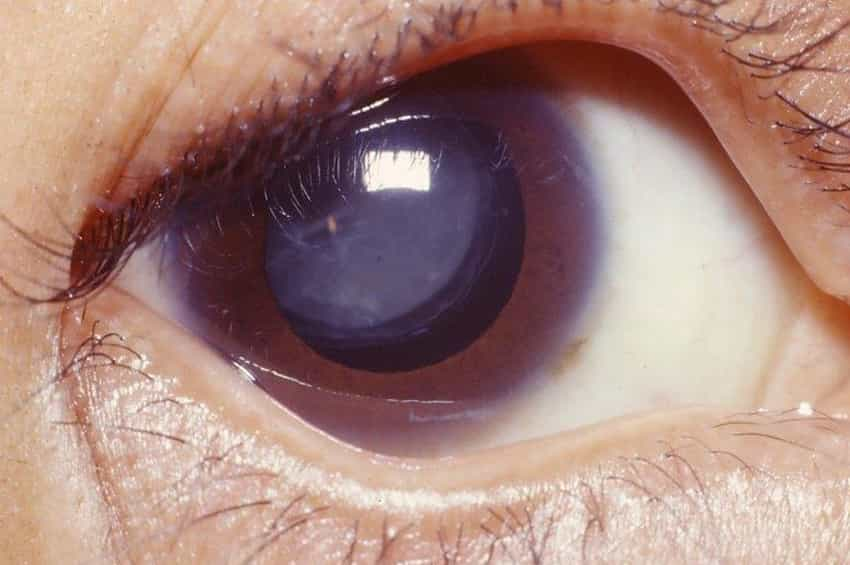
Teşhisi
Weill-Marchesani sendromunun teşhisi, kapsamlı bir klinik muayene, eksiksiz bir hasta ve aile öyküsü, karakteristik fiziksel bulguların tanımlanması ve çeşitli özel testlere dayanılarak konulabilir. Bunlar tipik olarak, gözlerin içini görüntülemek için bir aletin kullanılması (oftalmoskopi) gibi oküler muayeneleri içerir; göz içindeki basıncı ölçmek için teknikler (örn. tonometri); görme alanı testi; ve/veya diğer oküler teknikler.
Ek olarak, bozuklukla ilişkili olabilecek iskelet veya diğer anormallikleri saptamak ve karakterize etmek için gelişmiş görüntüleme teknikleri (örn. bilgisayarlı tomografi [BT] taraması veya manyetik rezonans görüntüleme [MRI]) veya diğer tanısal testler yapılabilir. CT taraması sırasında, iç yapıların enine kesit görüntülerini gösteren bir film oluşturmak için bir bilgisayar ve röntgen kullanılır.
Fiziksel bulgular otozomal resesif ve otozomal dominant Weill Marchesani sendromunu ayırt edemez. Otozomal resesif tipin tanısını doğrulamak için ADAMTS10 geni için moleküler genetik testler mevcuttur.
Tedavisi
Weill-Marchesani sendromunun tedavisi, her bireyde belirgin olan spesifik semptomlara yöneliktir. Bu tür bir tedavi, çocuk doktorları gibi tıp uzmanlarından oluşan bir ekibin koordineli çabalarını gerektirebilir; göz uzmanları (örneğin, oftalmologlar ve optometristler); iskelet, eklemler, kaslar ve ilgili dokuların bozukluklarını teşhis eden ve tedavi eden doktorlar (ortopedistler); ve kalp anormalliklerini teşhis eden doktorlar (kardiyologlar).
Weill-Marchesani sendromu için spesifik tedaviler semptomatik ve destekleyicidir. Uzmanlar, oküler anormalliklerin erken teşhisinin, optimal görsel gelişimin sağlanmasına yardımcı olmak için önemli olabileceğini belirtmektedir. Bazı durumlarda, görüşü iyileştirmeye yardımcı olmak için düzeltici gözlükler, diğer görsel yardımcılar ve/veya ameliyat önerilebilir.
Ek olarak, gözlerinde artan sıvı basıncı veya glokomu olanlar için tedavi, ilaçlı göz damlası tedavisi gibi göz içindeki basıncı (göz içi basıncı) kontrol etmeye yardımcı olacak önlemleri içerebilir; gözün renkli bölgesinde bir delik oluşturmak için lazer tedavisi (lazer iridektomi) veya irisin bir kısmının cerrahi olarak çıkarılması (iridotomi); merceğin çıkarılması; ve/veya diğer teknikler.
Uzmanlar, göz bebeklerinin uyarılmasının (miosis) veya genişlemesinin (midriyazis) etkilenen bazı kişilerde glokoma neden olabileceğini belirtiyor. Bu nedenle, göz bebeklerinin kasılmasına neden olan (yani kontrendike olan) ilaçlarla tedaviden kaçınılmalı ve gözlerin genişletilmesi son derece dikkatli yapılmalıdır.
Etkilenen kişiler, anestezi almadan önce doktorlarına bu teşhisi bildirmelidir. Eklem sertliği ve kraniyofasiyal anormallikler hava yolu yönetimini etkileyebilir. Weill-Marchesani sendromlu bireyler ve aileleri için genetik danışmanlık önerilmektedir. Bu bozukluğun diğer tedavisi semptomatik ve destekleyicidir.
Not: Sunulan bilgilerin amacı herhangi bir hastalığı teşhis veya tedavi etmek, iyileştirmek veya önlemek değildir. Tüm bilgiler yalnızca genel bilginize yöneliktir, tıbbi tavsiye veya belirli tıbbi durumların tedavisinin yerine geçmez. Uygulamadan önce bu bilgileri doktorunuzla görüşün.