Alfa-gal alerjisi ilk olarak 2009 yılında birkaç hastada tanımlandı. 5 yıl içinde bunların buzdağının sadece görünen kısmı olduğu ortaya çıktı. Durum, kanser tedavisinde kullanılan monoklonal antibiyotik setuksimaba karşı immünoglobulin E (IgE) aracılı bir alerjidir.
Haber Merkezi / Antikor, setuksimab molekülünün FAB kısmında yer alan alfa-gal adlı bir oligosakkarite bağlanır. Bu oligosakarit veya iki şekerli molekül, kimyasal olarak galaktoz-alfa-1,3-galaktoz olarak adlandırılır ve bu, iki galaktoz şeker molekülünün 1 ve 3 konumlarındaki karbon atomları arasındaki bir glikozit bağı ile bir arada tutulduğunu gösterir. Akut veya gecikmiş anafilaksi oluşturmak için alerjik hastaların vücutlarında önceden var olan IgE antikorları.
Bu garip reaksiyonun birincil nedeni, ABD’de Lone Star kene veya Amblyomma americanum adı verilen kene türlerinin ısırması iken , Fransa’da Ixodes ricinus ve Avustralya’da Ixodes holocyclus ısırmasıdır. Alfa-gal’e bağlanan bu tür IgE, alerjik rinit veya astım oluşturmayan bir alerjik reaksiyon örneğidir.
Alfa-gal alerjisi, proteinlerden ziyade etle ilişkili karbonhidratlara maruz kalmanın neden olduğu ve maruz kalma ile reaksiyon arasında saat olarak ölçülen bir zaman gecikmesiyle ortaya çıkan hücre aracılı alerjinin farklı bir türüdür. Gecikmiş tipteki alfa-gal alerjisinde, reaksiyonun gücünü etkileyen faktörler arasında doz, kene ısırmasına bağlı olarak maruz kalma süresi ve yenen etin türü yer alır.
Birçok hastada az miktarda hayvan eti aşırı duyarlılık reaksiyonu oluşturmaz. Bununla birlikte, daha yüksek dozlar alerjiyi hızlandırabilir ve bir tabak mangalda domuz eti gibi tam yardımlar genellikle çok sistemli bir anafilaktik reaksiyona neden olur. Daha yüksek yağ içeriğine sahip etler, hasta birkaç gün önce aynı tür et yağsız yemiş olsa bile, sıklıkla acil tedavi gerektiren ciddi reaksiyonlara neden olur.
Kene ısırmasından itibaren geçen süreye ilişkin zamanlama, IgE antikor üretiminin zamanla doğal olarak azaldığını, ancak tekrarlanan ısırıklarla yeniden etkinleştirildiğini düşündürmektedir. Bu nedenle, hastalar birkaç ay boyunca ete karşı herhangi bir reaksiyon göstermeyebilir, ancak daha sonra yakın zamanda bir kene ısırmasından sonra aniden şiddetli bir anafilaktik reaksiyon geliştirir.
Yemekten semptomların ortaya çıkmasına kadar geçen tipik gecikme yaklaşık 3-6 saattir. Bu, oligosakkaritin absorbe edilmesi ve 3-4 saat süren kan dolaşımına taşınması için gereken sürenin yanı sıra şilomikronlar üzerindeki alfa-gal antijenlerinin genel dolaşıma taşınması ile açıklanabilir.
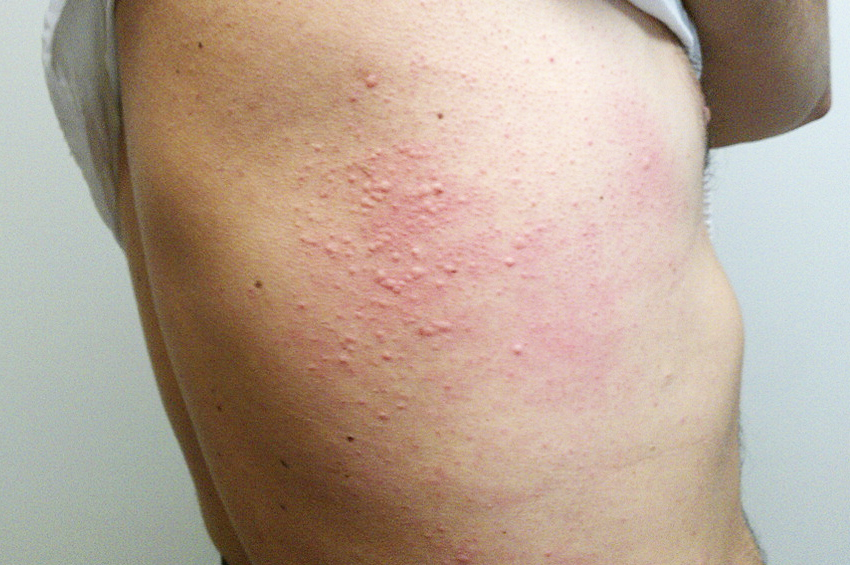
Belirtileri
Alfa-gal alerjisinin en yaygın semptomu, genellikle tüm vücutta, özellikle avuç içlerinde ve plantar deride şiddetli kaşıntıdır. Bu genellikle ürtiker ile ilişkili olabilir.
Diğerleri, alerjik reaksiyon başlamadan önce midelerinin bulandığını, ishal yaşadıklarını veya hazımsızlık yaşadıklarını bildirmiştir. Bu tür belirtiler bir gereklilik değildir ve birçok hastada alerjinin başladığı konusunda onları uyaran hiçbir belirti yoktur.
Önlemler
Alfa-gal alerjisi olan hastalar kırmızı etten kaçınmalıdır. Bazı durumlarda süt ürünleri diyete dahil edildiğinde semptomlar devam ederse sütten de kaçınmalıdırlar.
Tedavisi
Alfa-gal alerjisi, çoklu sistem ve ölümcül anafilaktik reaksiyon olasılığı nedeniyle acil olarak tedavi edilmelidir. Bu nedenle, reaksiyonu durdurmak için intramüsküler bir epinefrin enjeksiyonu gereklidir. Bu, alerjik belirtilerin altında yatan inflamatuar değişiklikleri hemen tersine çevirebilen tek ilaçtır ve anafilaksi vakalarında başka bir ilaçla değiştirilmemelidir. Yanıt yetersizse veya hasta ilaca dirençliyse tekrar dozlama gereklidir.
Bronkokonstriksiyonu temizlemek için albuterol gibi bronkodilatörler gereklidir. Anafilaksi sırasında hava yolu daralmasını önleyebilen bu beta reseptör agonistleri, nebulizasyon veya ölçülü doz inhaler yoluyla uygulanabilir.
Ek olarak, reaksiyon daha şiddetli ise oksijen uygulamasıyla birlikte kortikosteroidler ve antihistaminikler gerekebilir. Bu ilaçlar, alerjik inflamasyon sırasında inflamatuar aracıları serbest bırakan mast hücre ve bazofil degranülasyonunu önler.
Hipotansiyonu tedavi etmek için intravenöz sıvılara ek olarak başka vazopresör ilaçlar gerekebilir. Bradikardi varsa atropin ve yeterli kan şekeri düzeylerini sağlamak için glukagon gibi ilaçlar da verilebilir. Bu durum fark edildikten sonra, hastalar hızlı tedavi için yanlarında epinefrin (EpiPen) içeren bir otomatik enjeksiyon cihazı taşımalıdır.
Ek olarak, yiyeceklerin alınmasını takiben herhangi bir semptom gelişirse, hastalar derhal tıbbi yardım almalıdır. Bu kişilere ayrıca, antikor seviyeleri zamanla yavaş yavaş düştüğü için kene ısırıklarından kaçınmaları tavsiye edilir. Bu nedenle, Alfa-gal alerjisi olan bazı hastalar, birkaç yıl sonra kene ısırığı olmaksızın kırmızı eti tolere edebilirler.
Dikkat: Sayfa içeriği sadece bilgilendirme amaçlıdır.