İlaç döküntüleri, hastaneye yatırılan kişilerin yaklaşık %3’ünde meydana gelen ilaçlara karşı olumsuz cilt reaksiyonlarıdır. Deri hastalıklarına çok benzeyebilirler, ancak genellikle ilaç bırakıldığında kaybolurlar.
Haber Merkezi / Bazı ilaç döküntüleri yaşamı tehdit edebilir. Bu tür reaksiyonların ana özellikleri şunlardır;
- Yüz veya mukoza zarlarını içeren döküntüler
- Vücudun birçok yerinde birleşik döküntüler
- Kabarcıklar, büller, nekrotik veya purpurik lezyonlar
- Cilt ağrısı
- Boğaz veya dili içeren şişlik
- Yüksek ateş
- Lenf düğümü şişmesi
- Eklem ağrısı
- Kemik iliği, karaciğer veya böbrek parametrelerinde anormal değişiklik
- Solunum güçlükleri
- Kan basıncında anormal düşüş
En tehlikeli ilaç döküntüleri, toksik epidermal nekroz (TEN) ve ilaç aşırı duyarlılık sendromunu içerir.
Risk faktörleri
Aşağıdaki kategorilerde advers ilaç reaksiyonu insidansı daha yüksektir:
- 3 yaşından küçük erkekler
- 9 yaşından büyük kadınlar
- Genel olarak kadınlar
- Yaşlı hastalar
İlaç erüpsiyonu türleri
İlaç döküntüleri alerjik reaksiyonlara veya immün olmayan kutanöz reaksiyonlara bağlı olabilir. İlaçlara bağlı alerjik reaksiyonlar şunları içerir:
- Tip 1; Artan şiddet ölçeğinde ürtiker döküntüleri, anjiyoödem ve anafilaksi ile IgE aracılı bir reaksiyondur. Bir örnek insülindir.
- Tip 2; Bunlar sitotoksik reaksiyonlardır ve hemoliz ve purpura üretir. Bir örnek penisilin kaynaklı purpuradır.
- Tip 3; Bunlar, vaskülit, serum hastalığı ve ürtiker döküntülerine neden olan bağışıklık komplekslerinin oluşumundan kaynaklanır. Bir örnek salisilat kaynaklı serum hastalığıdır.
- Tip 4; Bunlar, maruziyetten bir ila üç hafta sonra meydana gelen ve hücre aracılı olan gecikmiş reaksiyonlardır. Genellikle topikal ilaçlar nedeniyle doza bağlı değildirler.
Alerjik olmayan ilaç döküntüleri, kendine özgü ilaç reaksiyonları, enzim eksiklikleri, irritan dermatit ve kümülatif toksisite nedeniyle olabilir.
Başlıca ilaç döküntü türleri
Aşağıdakiler başlıca ilaç reaksiyonlarıdır:
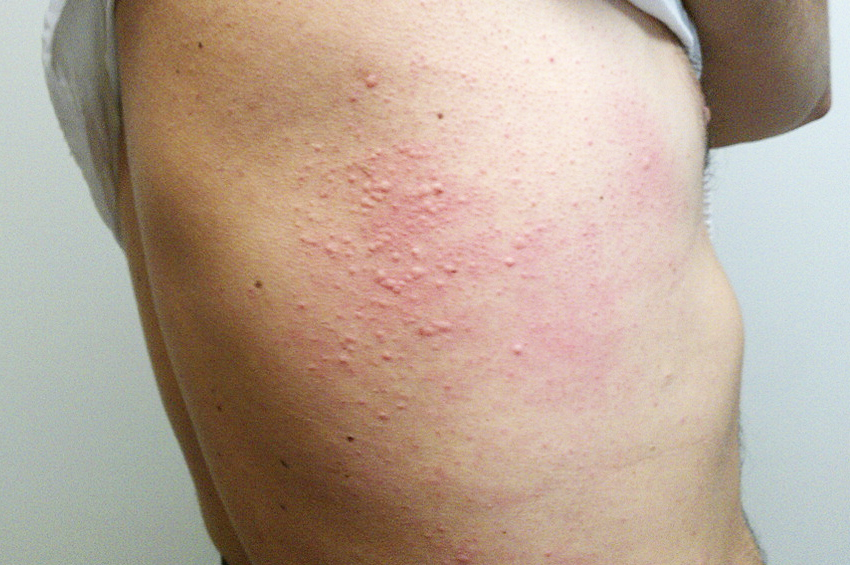
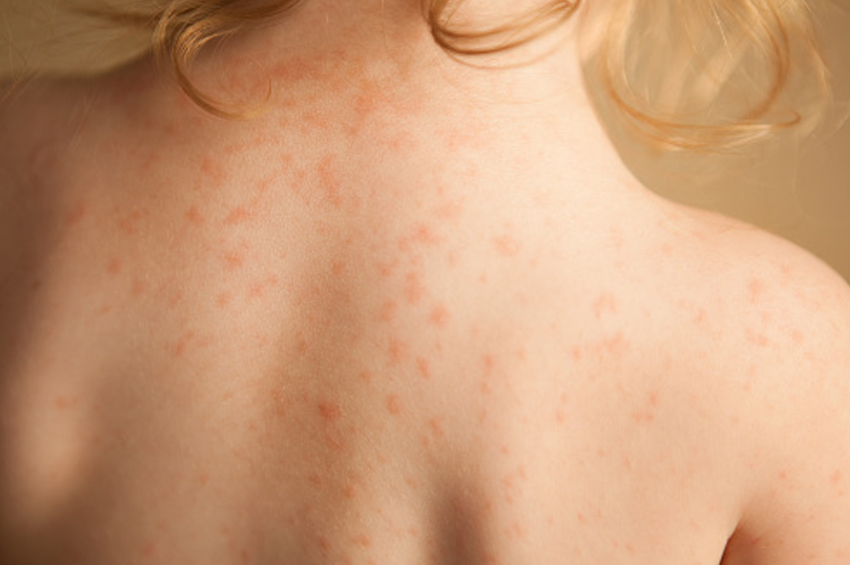
İlaç ekzantemleri: Bunlar genellikle yeni ilacın başlangıcından 5-10 gün sonra ortaya çıkar, ancak ikinci kez uygulandığında süre 1-3 gün kadar kısa olabilir. Genellikle simetrik maküller ve papüller (morbilliform) ile kendini gösterir, ancak kızıl hastalığına (scarlatiniform) benzeyebilir. TEN gibi ciddi reaksiyonlara ilerleyebilirler. Genellikle, rahatsız edici ilaç kesildiğinde bir hafta içinde azalırlar. Bazı yaygın örnekler NSAID’ler, penisilin ve sülfa ilaçlarıdır.
İlaca bağlı ürtiker: Bu, bazen dudak, dil veya boğazda şişme ile ilişkili cilt üzerinde kırmızımsı kabarcıklardan oluşur. İlk vaka ilaca maruz kaldıktan sonra 3 haftaya kadar ortaya çıkabilirken, tekrarlanan maruziyetler ürtikeri dakikalar içinde hızlandırabilir. Anafilaksi de oluşabilir.
Sorumlu ilaçlar çoğunlukla penisilin, opioidler ve NSAID’lerdir. Ürtiker, ateş ve eklem ağrısı ile birlikte, lenf nodu büyümesi ve endokardit gibi diğer semptomlarla birlikte veya bunlar olmadan ortaya çıktığında, buna serum hastalığı denir.
Sabit ilaç döküntüsü (FDE): Kişi aynı ilaca maruz kaldığında her zaman aynı noktada sabit bir ilaç püskürmesi meydana gelir; genellikle dudaklarda, ellerde, ayaklarda veya cinsel organlarda. Çözülmesi birkaç gün sürer ve arkasında mor bir iltihap sonrası renk değişikliği bırakır. Doksisiklin ve kotrimoksazol gibi ilaçlardan kaynaklanabilir. İlk maruziyetin ardından gelişmesi bir veya iki hafta sürerken, sonraki nüksler 48 saat içinde ortaya çıkar.

Purpurik döküntüler: Bunlara trombosit oluşumunu engelleyen ilaçlar gibi ilaçlara tepki olarak deriye kanama neden olur. Mekanizmalar, kılcal damarların artan kırılganlığını, alerjik trombositopeni ve kılcal iltihabı içerir.
İlaca bağlı ışığa duyarlılık: Bu, cildin açıkta kalan bölgelerinde güneş ışığının neden olduğu döküntüleri ifade eder, ancak güneş ışığına alışkın olan yüz ve kollar gibi alanlar hassasiyet belirtileri göstermeyebilir. Döküntü toksisite tarafından indüklenebilir, bu durumda ilacı kritik bir dozu aşan seviyelerde alan tüm kişilerde görülür ve güneş yanığı gibi görünecek kadar şiddetli olabilir. Bu, griseofulvin, tetrasiklinler ve NSAID’ler tarafından çökeltilir. Öte yandan, egzama veya likenoid reaksiyon şeklini alarak gerçekten alerjik olabilir. Tiyazidler, sülfonamidler ve fenotiyazinler bu tip reaksiyonlara neden olabilir.
İlaca bağlı hiperpigmentasyon: Bazı ilaçlar, melanin, hemosiderin veya bilinmeyen mekanizmalarla artan cilt rengi gibi çeşitli tiplerde pigment granüllerinin birikmesine neden olur. Bunlara amiodaron, klofazimin, demir veya minosiklin dahildir.
Akut jeneralize ekzantematöz püstüloz: Bu, eritematöz bir taban üzerinde çok sayıda yaygın steril püstüllerle işaretlenmiş ve 3 hafta kadar içinde düzelen bir döküntü türüdür. İlişkili ateş olabilir ve toplam lökosit ile eozinofil sayısı yüksektir. Suçlu ilaçlar, beta-laktamları ve eritromisin gibi makrolidleri içerir.
Eritema multiforme: Eritema multiforme hedef lezyonların varlığı ile karakterizedir ve enfeksiyonlara ve ilaç alerjilerine bağlı olarak sırasıyla minör ve majör formlar olarak sınıflandırılır. Mukoza zarları ayrıca kabarcık veya ülser oluşumunda rol oynar. Eritema multiforme’ye neden olabilen ilaçlar arasında sülfonamidler ve antikonvülzanlar bulunur.
İlaç aşırı duyarlılık sendromu (DHS): Buna eozinofili ve sistemik sendromlu (DRESS) ilaç döküntüsü de denir. Morbiliform bir döküntü, ateş ve iç organlarda hasar belirtilerini içeren ve yaklaşık %10’luk bir ölüm oranına sahip bir durumu ifade eder. Çoğu durumda, hastanın vücudunun bir kısmında şişme, püstüler döküntüler, lenf nodu büyümesi ve eozinofili vardır. Bu olduğunda, fulminan hepatit en yaygın ölüm nedenidir. Bu tür ilaçların yaygın örnekleri arasında fenitoin ve diğer antikonvülzanlar ve sülfonamidler bulunur.
Toksik epidermal nekroliz/Steven-Johnson sendromu: TEN, çoğunlukla sepsise bağlı olarak %30’dan fazla mortaliteye sahip, nadir fakat tehlikeli bir ilaç aşırı duyarlılık reaksiyonudur. Ateşle birlikte cildin %30’dan fazlasını tutan yaygın eritem özellikleri gösterir. Mukoza da tutulabilir. Hafif basınç altında yayılan kabarcıklar oluşur.
Eritem, deri ve mukozanın epidermal katmanlarının dökülmesini 3 gün içinde tamamlayacak şekilde ilerler. Cildin %10’dan azı tutulduğunda Steven-Johnson sendromu (SJS) denir ve ölüm oranı %5 civarındadır. Aradaki herhangi bir şey bir SJS/TEN örtüşmesidir.
Bu reaksiyondan sorumlu ilaçlar genellikle 1-3 hafta öncesinden başlatılmıştır. Aynı kategorideki tüm ilaçlarda olduğu gibi ilgili ilaçtan kaçınılmalıdır. Aile üyeleri de ilaç için test edilmelidir. TEN’e neden olan ilaçlar arasında; sülfonamidler, fenitoin
Teşhis ve tedavi
Hastanın kullandığı ilaçlardan birinin nedensel faktör olduğuna dair ciddi bir şüphe varsa, bir ilaç döküntüsü tanınır. Hastanın son 12 hafta içinde almış olduğu tüm ilaçlar dikkate alınmalıdır. İlaç döküntüleri için spesifik bir kan testi yoktur. Ancak tam kan sayımı, kanda eozinofili varlığını ortaya çıkarabilir.
Kapsamlı cilt tutulumu durumunda veya lezyonlar likenplanus veya sedef hastalığı gibi diğer cilt hastalıklarını taklit ediyorsa, gerekli olabilecek diğer testler arasında cilt biyopsisi bulunur. Hepatorenal fonksiyonun da değerlendirilmesi gerekebilir.
Bir ilacın bir hastada alerjiye neden olduğundan şüphelenildiğinde, ilacın sınıfı ve hasta çizelgesinde ve taburcu notlarında belirtilen spesifik ilaç kesilmeli ve gelecekte o sınıftan kaçınılmalıdır. Lokal ilaç döküntülerinin tedavisi topikal steroid merhemler, iltihaplı cildi yatıştırmak için yumuşatıcılar ve kaşıntıyı hafifletmek için antihistaminikler ile yapılır. Sistemik reaksiyonlar veya Steven-Johnson sendromunun hastaneye yatırılması ve sıklıkla yoğun bakıma ihtiyacı vardır. Hastanın her zaman ilacın adını ve tepkilerini taşıyan bir sağlık kartı olmalıdır.
Dikkat: Sayfa içeriği sadece bilgilendirme amaçlıdır.